Life Technologies Japan Ltd.
- HOME
- Life Technologies Japan Ltd.
- Product & Service
- From precious samples to precise insights with the Sequential Protein/DNA/RNA Extraction Kit
Business Category : Materials for research and development
Main Service Region :
Product & Service
From precious samples to precise insights with the Sequential Protein/DNA/RNA Extraction Kit
Service Category :Analysis、Development、Modality
Sample prep
From precious samples to precise insights with the Sequential Protein/DNA/RNA Extraction Kit
A bead-based multiomics solution
| Authors Marie Holter-Sørensen, Samrawit Michael, Iwona Grad, Laure Jobert, Charmaine Hinahon, Leigh Foster, Lina Zheng, Ketil Winther Pedersen, and Berit Marie Reed, Thermo Fisher Scientific. |
Introduction
Multiomics refers to the integrated analysis of multiple molecular components—such as proteins, DNA, and RNA—to obtain a comprehensive view of biological systems and their regulatory networks. It facilitates biomarker discovery, advances precision medicine, and accelerates liquid biopsy research, among many additional applications. Because a single omics layer captures only one dimension of biological regulation, integrating proteomics, genomics, and transcriptomics enables a more comprehensive, system-level characterization of an organism’s molecular state. Multiomics applications increasingly require matched protein, DNA, and RNA from the same biological specimen in order to preserve analyte consistency, increase biological insight, and reduce variability, but also because the sample is limited [1]. Traditional extraction workflows often require splitting specimens and performing separate protein, DNA, and RNA isolations, increasing variability and risking loss of analyte concordance. These approaches also consume substantial amounts of precious or limited samples, making it challenging to generate complete multi-analyte datasets. In addition, traditional column-based methods are manual and time-intensive, limiting throughput and increasing hands-on effort.
The Thermo Scientific™ Sequential Protein/DNA/RNA Extraction Kit addresses these challenges by enabling all three analyte categories to be isolated sequentially from a single lysate, maximizing data yield while minimizing sample consumption. The automationready magnetic-bead workflow reduces hands-on time and supports scalable, highthroughput processing. The integrated workflow preserves sample integrity and increases recovery from challenging, heterogeneous, and limited specimens, with multi-analyte extracts suitable for downstream mass spectrometry, gel electrophoresis–based applications including western blotting, and next-generation sequencing, qPCR, and RT-qPCR. Unlike column-based systems, this sequential extraction kit incorporates a dual magnetic bead system comprising Invitrogen™ Dynabeads™ magnetic beads for protein handling and Applied Biosystems™ MagMAX™ magnetic beads for DNA and RNA recovery, offering high reproducibility and an easy-to-use, automation-friendly solution.
The Sequential Protein/DNA/RNA Extraction Kit was developed for processing a wide variety of sample types obtained from limited specimens, including 100,000 cells or 1 mg of tissue, with flexibility to handle up to 1,000,000 cells or 10 mg of tissue.
Moreover, the kit enables efficient extraction from precious and extremely small samples, such as 1,000 cells or 5 µg of tissue. The kit supports both manual and automated workflows on Thermo Scientific™ KingFisher™ sample purification systems.
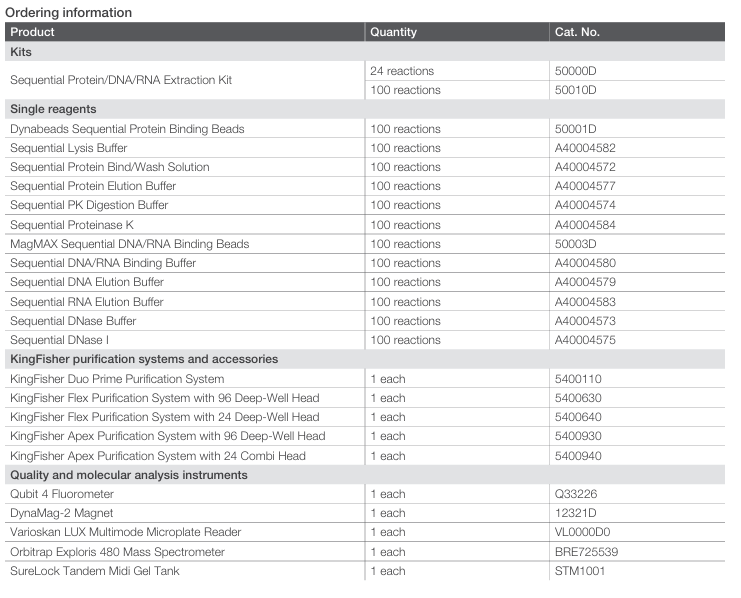
When used on KingFisher instruments, up to 96 sequential extractions of protein, DNA, and RNA can be completed in approximately 2.5 hours from start to finish. Figure 1 shows the automated sequential isolation workflow, expected touch points, and total processing time. Additional information is available in the kit’s user guide, and protocols for both manual and automated workflows for KingFisher systems, along with automation scripts, can be found at thermofisher.com/order/catalog/product/50000D.
A new spatial biology is in sight—literally. It is a more vivid, more detailed, and ultimately more informative spatial biology. It can distinguish between cell types that were once indistinguishable, and it can do so while capturing their spatial context. It can even pinpoint the subcellular locations of individual molecules. And it can accomplish these tasks with unprecedented precision because technology is now available that opens a new dimension beyond the usual three spatial dimensions. This new dimension may be called the “plex” dimension.
Plex refers to the number of fluorescence markers that are used with microscopy and other cell analysis platforms. Conventional platforms may accommodate just a handful of markers, constraining investigations of complex biological phenomena. But such investigations may require many markers.
Unfortunately, using more and more markers—and thereby shifting from low-plex to high-plex spatial biology—has been too difficult for most laboratories. They’ve balked at the need for special expertise, complicated workflows, and instrument upgrades. Fortunately, these difficulties can be overcome with new multiplex imaging technologies. For example, there are antibody panels that are compatible with streamlined workflows and automated imaging systems.
To learn more about these technologies, consult the articles in this eBook—especially the article describing organ mapping antibody panels. Also, be sure to read the articles that describe the kinds of spatial biology applications that are bound to become more common as high-plex technology becomes more accessible. Indeed, this technology is democratizing spatial biology.
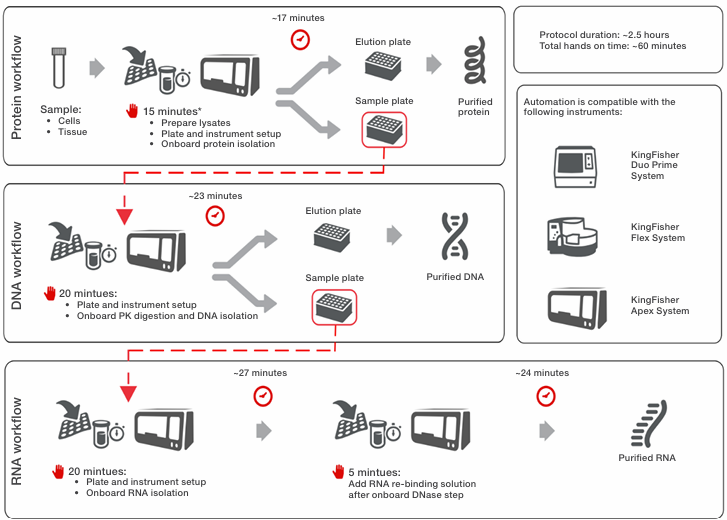
*Sample addition time may vary depending on the number of samples and method used to add samples to the sample plate
Figure 1. Schematic presentation of protein and nucleic acid extraction workflow using the Sequential Protein/DNA/RNA Extraction Kit on
KingFisher purification systems. Up to 96 protein, DNA, and RNA extractions can be completed within ~2.5 hours, including hands-on time.
Materials and methods
Protein and nucleic acid isolation HeLa cells were cultured in standard conditions. Peripheral blood mononuclear cells (PBMCs) originating from buffy coats from healthy human donors were either isolated or obtained commercially. Frozen liver tissue from mice, treated with Invitrogen™ RNAlater™ Stabilization Solution, was obtained commercially. 100,000 HeLa cells, 500,000 PBMCs, and 1–2 mg tissue were used for low sample inputs; for high sample inputs, 1,000,000 HeLa cells and 10 mg tissue were used. Testing of workflow scalability and sample input limits were achieved using serial dilution of lysates (HeLa cells and tissue) or cells (PBMCs). For HeLa cells, the lysate started from 100,000 cells per sample and was diluted down to 1,000 cells per sample. For tissue, the lysate started from 2 mg of tissue per sample and was serially diluted down to 0.005 mg of tissue per sample. For PBMCs, starting with 250,000 cells per sample, the cells were two-fold serially diluted down to ~8,000 cells per sample, centrifuged, and then lysed. Protein, DNA, and RNA from the three sample types were extracted on the KingFisher Flex, Apex, and Duo Prime purification systems or with a manual workflow using thermal mixers according to the user guide. Automated extractions were performed in 96 deep-well plates. The manual workflows were performed in 1.5 mL tubes using the Invitrogen™ DynaMag™-2 Magnet. To evaluate the reproducibility of the Sequential Protein/DNA/RNA Extraction Kit, extractions were done using different lots of the reagents.
Yield and quality
Extracted proteins were quantified using the Thermo Scientific™ Pierce™ BCA Protein Assay Kit and the Thermo Scientific™ Varioskan™ LUX Multimode Microplate Reader, and subsequently analyzed by SDS-PAGE followed by Coomassie staining. Extracted DNA and RNA yields and concentrations were analyzed utilizing the Invitrogen™ Qubit™ 1X dsDNA Broad Range/High Sensitivity Assay Kit and Qubit™ RNA Broad Range/High Sensitivity Assay Kit, respectively, on the Qubit™ 4 Fluorometer. DNA and RNA integrity were measured using Agilent™ genomic DNA and Agilent™ RNA reagents on the Agilent™ 4150 TapeStation System.
SDS-PAGE and Coomassie staining
Total protein extracts were analyzed by SDS-PAGE using Invitrogen™ Bolt™ Bis-Tris Plus Mini Protein Gels, with 10 µg of protein loaded per lane. Proteins were isolated from 100,000 HeLa cells, 1,000,000 PBMCs, and 1 mg liver tissue using the Sequential Protein/DNA/RNA Extraction Kit, RIPA lysis buffer, or Thermo Scientific™ T-PER™ Tissue Protein Extraction Reagent, as indicated. For liver samples, 1 mg of tissue was processed using the Sequential Protein/DNA/RNA Extraction Kit, while a comparative extraction was performed using 1 mg of liver tissuelysed per 40 µL of T-PER reagent. Samples were prepared with 4X LDS sample buffer supplemented with DTT and resolved alongside the Invitrogen™ SeeBlue™ Plus2 Pre-stained Protein Standard. Following electrophoretic separation, gels were stained with Thermo Scientific™ PageBlue™ Protein Staining Solution to visualize total protein profiles.
Mass spectrometry
Proteins were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using the Thermo Scientific™ Orbitrap Exploris™ 480 Mass Spectrometer. Protein samples obtained with the Sequential Protein/DNA/RNA Extraction Kit were further processed using the Thermo Scientific™ EasyPep™ Micro MS Sample Prep Kit, omitting the lysis step. The undiluted eluted sample was used to quantitate the peptide concentration with the Thermo Scientific™ Pierce™ Quantitative Colorimetric Peptide Assay Kit, and injection on the mass spectrometer was normalized to 500 ng per sample. Data analysis was carried out using Thermo Scientific™ Proteome Discoverer™ 3.1 Software. The workflow of the EasyPep Micro MS Sample Prep Kit served as the reference method.
Whole-genome sequencing
Libraries were prepared using the tagmentation-based and PCR based Illumina™ DNA Prep Kit and custom IDT™ 10 bp unique dual indices (UDI) with a target insert size of 280 bp. No additional DNA fragmentation or size selection steps were performed. Sequencing was performed on an Illumina™ NovaSeq™ X Plus sequencer in one or more multiplexed shared-flow-cell runs, producing 2 x 151 bp paired-end reads. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert1 (v4.2.4). Raw reads underwent quality control with the FastQC tool (bioinformatics.babraham.ac.uk/projects/fastqc/) and trimming with the Trim Galore tool (bioinformatics.babraham.ac.uk/projects/trim_galore/). The remaining reads were then aligned to the GRCh37 (hg19) assembly for HeLa cells and PBMCs and GRCm38 (mm10) assembly for mouse tissue with the BWA tool (pubmed.ncbi.nlm.nih.gov/20080505/). High mapping quality and paired reads were retained and further deduplicated by Picard tools (broadinstitute.github.io/picard/), and then the uniformity metric was calculated per sample. For use as a reference, DNA was isolated in parallel using the Thermo Scientific™ MagMAX™ DNA Multi-Sample Ultra 2.0 Kit.
rRNA depletion sequencing
Samples were DNase-treated with Invitrogen™ DNase (RNase free). Library preparation was performed using the Illumina™ Stranded Total RNA Prep Ligation with Ribo-Zero Plus kit and 10 bp unique dual indices (UDI). Sequencing was done on a NovaSeq X Plus sequencer, producing paired-end 150 bp reads. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert (v4.2.4). Raw reads underwent quality control with the FastQC tool (bioinformatics.babraham.ac.uk/projects/fastqc/) and trimming with the Trim Galore tool (bioinformatics.babraham.ac.uk/projects/trim_galore/). The remaining reads were then aligned to the GRCh37 (hg19) assembly for HeLa cells and PBMCs and GRCm38 (mm10) assembly for mouse tissue with STAR software (pubmed.ncbi.nlm.nih.gov/23104886/). Gene expression read counts were identified using the featureCounts program (academic.oup.com/
bioinformatics/article/30/7/923/232889) with standard filtering criteria. For use as a reference, RNA was isolated in parallel using the Applied Biosystems™ MagMAX™ mirVana™ Total RNA Isolation Kit.
MicroRNA detection
Total RNA was extracted from 100,000 HeLa cells using the Sequential Protein/DNA/RNA Extraction Kit or the MagMAX mirVana Total RNA Isolation Kit as a reference. cDNA was generated using the Applied Biosystems™ TaqMan™ MicroRNA Reverse Transcription Kit, and miR-16 and let-7a levels were quantified by real-time PCR using Applied Biosystems™ TaqMan™ miRNA assays. Relative miRNA expression was compared between the two extraction methods, with the mirVana kit serving as the control to confirm that RNA was not lost during the sequential extraction process. RNA extraction was performed in duplicate, and RT-PCR was conducted in triplicate for each isolated RNA sample.
qPCR/RT-qPCR
DNA and RNA were extracted from decreasing amounts of cells and tissue using the Sequential Protein/DNA/RNA Extraction Kit. One-tenth of the extracted DNA samples (5 µL) were used as template in real-time PCR using Applied Biosystems™ TaqMan™ Assays. TaqMan Assays detecting GAPDH genomic DNA corresponding to the species were used. One-tenth of the extracted RNA samples (5 µL) were used as template, reversed transcribed, and then directly quantified by real-time PCR using the Invitrogen™ EXPRESS One-Step SuperScript™ qRT-PCR Kit. cDNA-specific TaqMan Assays detecting ACTB (HeLa cells) or HPRT (mouse liver tissue) were used. qPCR was performed on the Applied Biosystems™ QuantStudio™ 5 Real-Time PCR System.
Results and discussion
Consistent yields and integrity across small and various sample types
As no single omics layer can provide a comprehensive view of biological systems, combining multiple omics modalities is essential for integrative, system-level understanding and more accurate interpretation of disease biology. Traditional workflows require splitting a sample to isolate protein, DNA, and RNA
separately, whereas the Sequential Protein/DNA/RNA Extraction Kit enables sequential recovery of all three analyte categories—protein, DNA, and RNA—from the same sample (Figure 2). For HeLa cells, PBMCs, and tissue samples, the recovered protein, DNA, and RNA yields fall well within the expected ranges for the respective starting material amounts, demonstrating efficient and consistent extraction across diverse sample types.
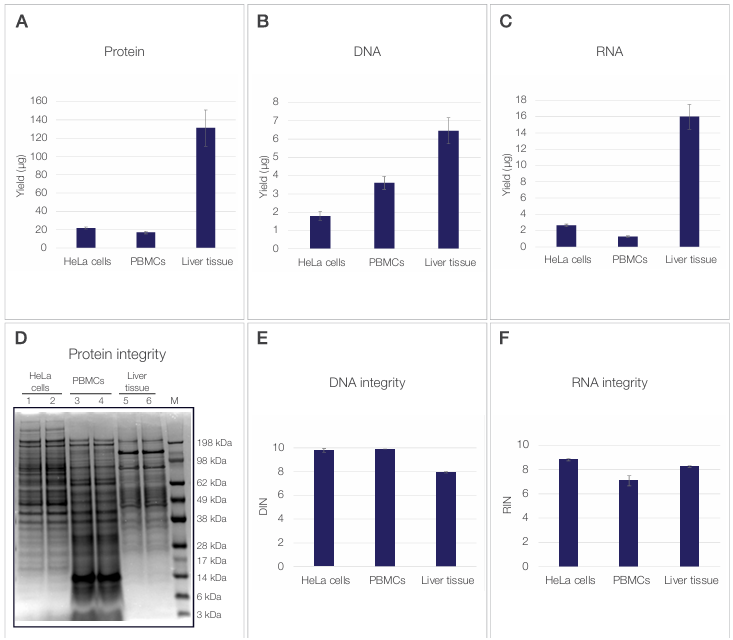
Figure 2. Protein, DNA, and RNA yield and integrity. (A) Protein, (B) DNA, and (C) RNA yields (µg) obtained from 100,000 HeLa cells, 500,000 PBMCs,
and 2 mg mouse liver tissue are shown as the mean of three independent reagent lots. Error bars represent the standard deviation between lots. (D) Protein
integrity was assessed by SDS-PAGE of total protein extracts visualized by Coomassie staining, with 10 µg of protein loaded per lane. Lanes 1 (HeLa cells),
3 (PBMCs), and 5 (liver tissue) contain protein extracts prepared using the Sequential Protein/DNA/RNA Extraction Kit. Lanes 2 (HeLa cells) and 4 (PBMCs)
contain protein extracts prepared using RIPA lysis buffer, and lane 6 (liver tissue) contains protein extracted using T-PER reagent. M indicates the molecular
weight marker (kDa). (E) DNA integrity and (F) RNA integrity were evaluated using DNA Integrity Number (DIN) and RNA Integrity Number (RIN), respectively,
from the same sample inputs. Values are shown as the mean of three independent reagent lots, with error bars representing the standard deviation.
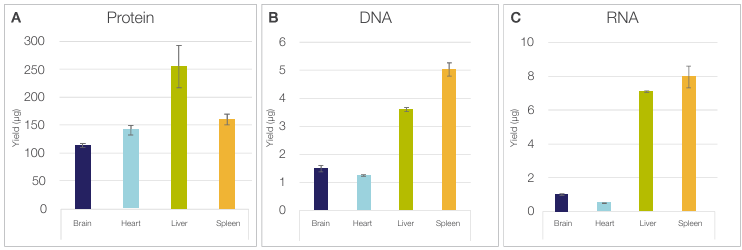
Figure 3. The Sequential Protein/DNA/RNA Extraction Kit is compatible with various tissues. Yields of (A) protein, (B) DNA, and (C) RNA obtained
from 1 mg of tissue across four different tissue types (brain, heart, liver, and spleen) are shown. Error bars represent the standard deviations across technical
replicates (n = 3).
Multi-tissue compatibility
Isolating sufficient quantities of protein, DNA, and RNA from diverse tissue types is often challenging due to their distinct biochemical and structural properties [2]. Brain tissue is highly enriched in lipids, which interfere with protein and nucleic acid solubilization and can reduce effective analyte recovery [3]. Heart tissue contains extensive extracellular matrix and relatively low nuclear density, limiting nucleic acid yield per unit mass [4]. Liver tissue exhibits high endogenous nuclease and protease activity, leading to rapid degradation of RNA, DNA, and proteins if not processed efficiently [5]. Spleen tissue contains abundant RNases and DNases that can compromise analyte integrity [6]. Despite these inherent challenges, the Sequential Protein/DNA/RNA Extraction Kit enables robust multi-analyte isolation from as little as 1 mg of each tissue type, allowing consistently high-quality protein, DNA, and RNA, as illustrated in Figure 3. Across all tissue types, DNA and RNA integrity remained high, with DIN and RIN values ≥7.0 for all samples (data not shown), confirming suitability for sensitive downstream analyses despite tissue heterogeneity and small sample size. Protein yields were consistent with known protein content of the tested organs, with brain producing the lowest yield and liver the highest. Similarly, DNA yield reflected expected tissue biology, including the high cell density of spleen and the polyploidy of liver. RNA yield aligned with the high metabolic activity of liver and the lymphocyte-rich nature of spleen.
Compatibility with mass spectrometry
To assess the proteome coverage and quality of protein recovery achieved with the Sequential Protein/DNA/RNA Extraction Kit, mass spectrometry (MS) analysis was performed on protein eluates obtained from 300,000 HeLa cells, 500,000 PBMCs, and 1–2 mg of mouse liver tissue. Across all sample types, MS data from proteins isolated using the Sequential Protein/DNA/RNA Extraction Kit showed chromatogram intensities comparable to those obtained with the reference method, as illustrated in Figure 4A–C (red vs. black chromatograms). Protein identification overlap remained high, with minimal differences between the Sequential Protein/DNA/RNA Extraction Kit and the reference workflow: 3.61% for HeLa cells, 6.35% for PBMCs, and 9.71% for mouse liver tissue, as shown in the Venn diagrams in Figure 4D–F. This high concordance in protein identifications is well within the typical variability observed in discovery proteomics workflows [7]. These small differences indicate that the sequential extraction method helps provide proteome coverage equivalent to the reference method. Additionally, reproducibility across multiple kit lots demonstrated the robustness of the workflow for proteomics applications with coefficients of variation of <5.2% (data not shown). Together, these results demonstrate that the Sequential Protein/DNA/RNA Extraction Kit enables efficient extraction of a broad protein range from both cells and tissues, yielding protein samples that are fully compatible with and well suited for high
quality mass spectrometry–based proteomic analysis.
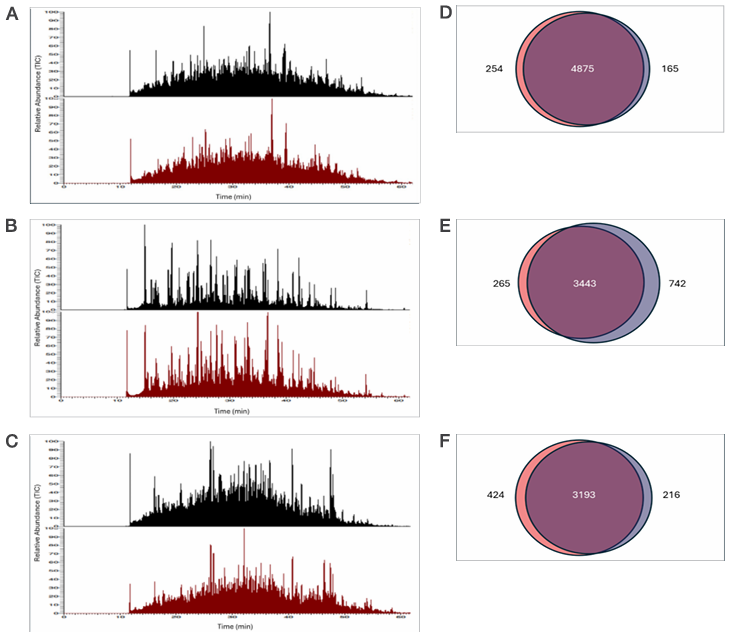
Figure 4. Compatibility of isolated proteins with mass spectrometry. Chromatograms represent proteins extracted from (A) 300,000 HeLa cells, (B)
500,000 PBMCs, and (C) 2 mg mouse liver tissue using either the Sequential Protein/DNA/RNA Extraction Kit (red) or the reference method, the EasyPep
Micro MS Sample Prep Kit (black). Venn diagrams on the right show comparisons of proteins identified from (D) HeLa cells, (E) PBMCs, and (F) mouse liver
tissue using the sequential kit (blue) and the EasyPep kit (pink). Proteins identified by both workflows are shown in purple.
Compatibility with whole-genome sequencing
Genomic DNA isolated using the Sequential Protein/DNA/RNA Extraction Kit was used for whole-genome sequencing across multiple sample types. DNA was extracted from 300,000 HeLa cells, 500,000 PBMCs, and 1–2 mg mouse liver tissue preserved in RNAlater solution and sequenced on an Illumina platform. As shown in Figure 5A, genome-wide read coverage profiles generated from sequentially extracted gDNA closely matched those obtained using a reference extraction method, demonstrating consistent genome-wide coverage across all sample types. In addition, sequencing performance metrics—including total read output, uniquely mapping reads (Q30), mean sequencing depth, and coverage uniformity—were comparable between the sequential and reference extraction methods (Figure 5B).
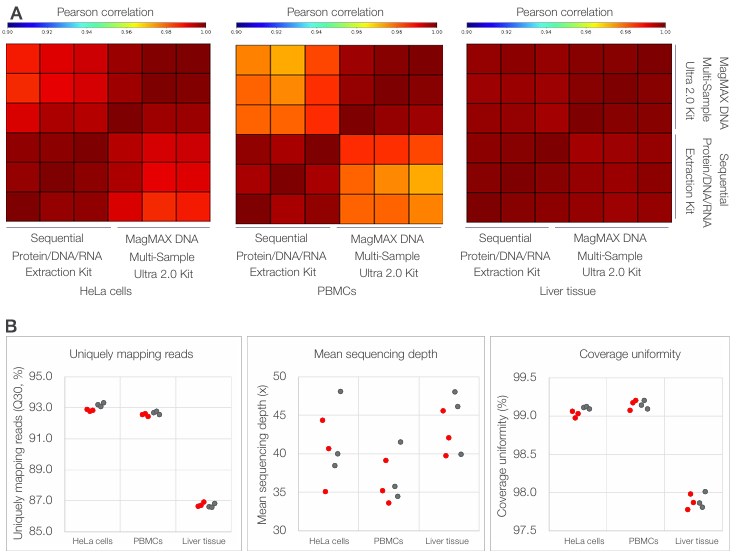
Figure 5. Compatibility of sequentially extracted gDNA with whole-genome sequencing across diverse sample types. (A) Genome-wide coverage
correlation heatmaps showing pairwise Pearson correlation coefficients of normalized read depth calculated in nonoverlapping 50 kb bins for HeLa cells,
PBMCs, and mouse liver tissue. Heatmaps compare gDNA isolated using the Sequential Protein/DNA/RNA Extraction Kit with gDNA isolated using the
MagMAX DNA Multi-Sample Ultra 2.0 Kit. (B) Summary of whole-genome sequencing performance metrics, including uniquely mapping reads, mean
sequencing depth, and coverage uniformity, for sequentially extracted gDNA (red) and reference-extracted gDNA (gray). Each point represents an
individual replicate (n = 3 per method and sample type).
Compatibility with rRNA depletion sequencing
Total RNA isolated using the Sequential Protein/DNA/RNA Extraction Kit was evaluated for compatibility with rRNA depletion RNA sequencing. RNA eluates were generated from 300,000 HeLa cells, 500,000 PBMCs, and 1–2 mg mouse liver tissue preserved in RNAlater solution, and libraries were prepared using an rRNA depletion workflow. Gene expression profiles derived from RNA isolated using the Sequential Protein/DNA/RNA Extraction Kit showed strong concordance with those obtained using the MagMAX mirVana Total RNA Isolation Kit across all sample types (Figure 6A). Analysis of gene detection overlap further showed that similarity between the sequential and reference extraction methods was comparable to replicate to-replicate similarity observed within each method, indicating that inter-method variability was within the range of intra-method variability (Figure 6B, left). Overall gene detection breadth and uniquely mapped read percentages were also comparable between the two extraction methods, demonstrating consistent RNA sequencing performance following rRNA depletion (Figure 6B, middle and right).
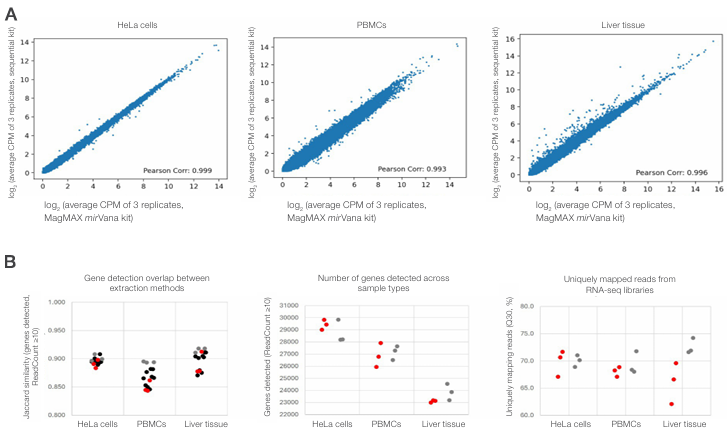
Figure 6. Compatibility of sequentially extracted total RNA with rRNA depletion sequencing across diverse sample types. (A) Scatter plots
showing gene expression concordance between RNA isolated using the Sequential Protein/DNA/RNA Extraction Kit and the MagMAX mirVana Total
RNA Isolation Kit for HeLa cells, PBMCs, and mouse liver tissue. Plots show log2-transformed average CPM values for each gene. (B) Summary of RNA
sequencing performance metrics comparing the two extraction methods. Left: gene detection similarity assessed by Jaccard similarity (genes detected,
ReadCount ≥10), showing replicate-to-replicate comparisons within the sequential method (red), within the reference method (gray), and between
methods (black). Middle: number of genes detected (ReadCount ≥10). Right: percentage of uniquely mapped reads (Q30). In the middle and right panels,
red and gray indicate data from the sequential and reference methods, respectively; points represent individual replicates.
A flexible solution for cells and tissue samples
Extraction of protein, DNA, and RNA from small samples of various biological materials using the automated workflow of the Sequential Protein/DNA/RNA Extraction Kit and KingFisher purification system provided excellent yields, high analyte integrity, and quality suitable for reliable downstream analysis. To evaluate the ability of the workflow to accommodate larger sample sizes, protein, DNA, and RNA were extracted from either 100,000 or 1,000,000 HeLa cells, using the same buffer and bead volumes. In parallel, the manual protocol was tested to confirm that the extraction can be performed just as efficiently manually when required. As expected, 1,000,000 HeLa cells yielded increased amounts of both protein and nucleic acids (Figure 7). Extraction efficiencies for each analyte fraction were comparable between the manual and automated workflows (Figure 7, light and dark blue bars). DNA and RNA integrity remained high for all samples, with DIN and RIN values ≥9.2 (data not shown). Together, these results demonstrate the flexibility, versatility, and robustness of the sequential extraction workflow.
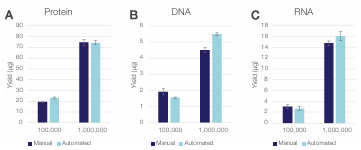 Figure 7. Flexibility of the Sequential Protein/DNA/RNA Kit for
Figure 7. Flexibility of the Sequential Protein/DNA/RNA Kit for
different sample sizes. (A) Protein, (B) DNA, and (C) RNA yields obtained
using manual vs. automated workflows for low (100,000) and high
(1,000,000) sample inputs of HeLa cells. The error bars represent standard
deviations of three technical replicates (n = 3).
miRNA retrieval
MicroRNAs (miRNAs) are short, noncoding RNA molecules that regulate gene expression by binding to target mRNAs and controlling their stability or translation [8]. As key post transcriptional regulators, they shape diverse cellular pathways by fine-tuning gene expression networks and can also influence epigenetic states indirectly through modulation of enzymes involved in chromatin regulation [9]. Their dysregulation is associated with both solid tumors and hematologic malignancies, where they may function as oncogenes or tumor suppressors [10]. The stability of circulating miRNAs in body fluids further supports their utility as minimally invasive cancer biomarkers [11]. Therefore, it is important that multiomics workflows efficiently recover short RNA species such as miRNAs. Examples of well characterized miRNAs include members of the let-7 family and miR-16. Dysregulation of let-7 can impair differentiation pathways [12], while miR-16 modulates leukemic cell survival by targeting apoptotic regulators such as BCL2 [13].
Both let-7 miRNAs and miR-16 were detected in RNA isolated from HeLa cells using either the Sequential Protein/DNA/RNA Extraction Kit or the MagMAX mirVana Total RNA Isolation Kit, which is designed for efficient capture of small RNAs. Detection using TaqMan miRNA assays showed comparable Ct values between the two extraction methods, demonstrating efficient recovery of miRNAs with both kits (Figure 8).
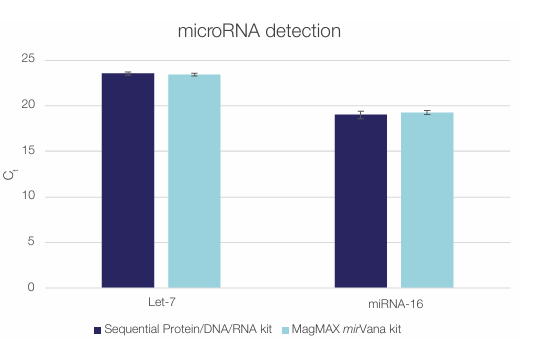 Figure 8. The Sequential Protein/DNA/RNA Extraction Kit efficiently
Figure 8. The Sequential Protein/DNA/RNA Extraction Kit efficiently
isolates miRNAs. Let-7 and miRNA-16 were detected in total RNA isolated
from 100,000 HeLa cells using the Sequential Protein/DNA/RNA Extraction
Kit (dark blue) or the MagMAX mirVana Total RNA Isolation Kit (light blue).
Error bars represent standard deviations of two technical replicates (n = 2).
Limited sample
When working with limited biological material, maximizing extraction efficiency becomes essential. Ultra-small samples—including needle biopsies, fine-needle aspirates, organoids, microdissected tissue regions, circulating tumor cell (CTC)-derived pellets, and rare primary cells—are increasingly common in translational research, oncology, and precision medicine. These materials often yield only a few micrograms of tissue or a few thousand cells, making it impractical to split the sample across separate protein, DNA, and RNA workflows without losing critical biological signal. A sequential multi-analyte isolation method overcomes this barrier by enabling multiomic profiling from a single input. The Sequential Protein/DNA/RNA Extraction Kit was evaluated using ultra-small inputs, including as few as 1,000 HeLa cells, 8,000 PBMCs, and as little as 5 µg of mouse liver tissue. Protein, DNA, and RNA were recovered from nearly all input samples, with yields decreasing proportionally with input size (Figure 9). RNA was not detected in the eluate from 8,000 PBMCs, most likely due to the limit of detection of the Qubit assay rather than failure of extraction. These results confirm that the Sequential Protein/DNA/RNA Extraction Kit can efficiently isolate protein, DNA, and RNA from ultra-small sample inputs.
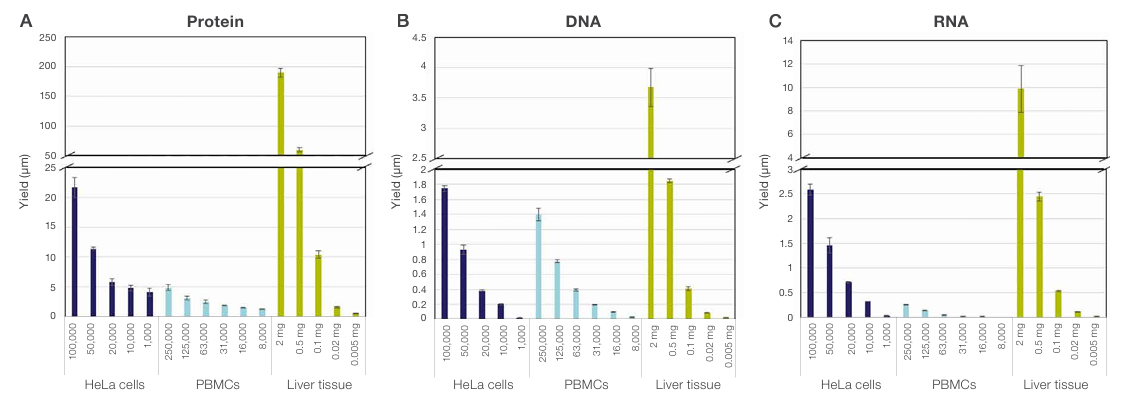
Figure 9. The Sequential Protein/DNA/RNA Extraction Kit enables isolation of protein, DNA, and RNA from ultra-small samples. Yields of (A)
protein, (B) DNA, and (C) RNA were obtained from serial dilutions of PBMCs or serial dilution of lysates of HeLa cells and mouse liver tissue.
The limited amounts of DNA and RNA obtained from ultra-low sample inputs do not allow robust assessment of nucleic acid quality using standard methods. To confirm that DNA and RNA extracted from such small inputs are compatible with downstream molecular analyses, qPCR and RT-qPCR were performed on nucleic acids isolated with the Sequential Protein/DNA/RNA Extraction Kit from progressively decreasing sample sizes down to 1,000 HeLa cells and 2.5 µg of mouse liver tissue.
Strong qPCR and RT-qPCR signals, even at the lowest sample input (Figure 10), demonstrated the robustness of the workflow and its ability to efficiently extract high-quality nucleic acids even from ultra-low inputs. The obtained Ct values closely matched those expected based on the dilution series of the starting material, confirming the linear scalability of the extraction workflow (Figure 10).
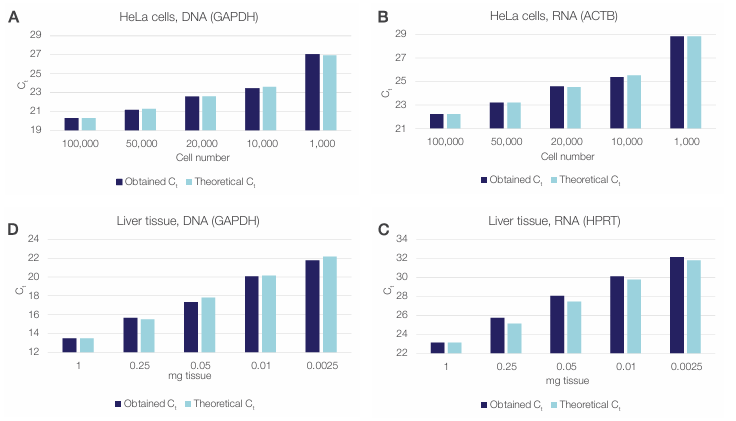
Figure 10. The Sequential Protein/DNA/RNA Extraction Kit enables isolation of functional DNA and RNA from ultra-small samples. The extracted
DNA and RNA was quantified using qPCR and RT-qPCR. Obtained Ct values (dark blue) for HeLa cell (A) DNA and (B) RNA and liver tissue (C) DNA and
(D) RNA are compared to the theoretical Ct values calculated from dilutions (light blue).
Robust workflow with high reproducibility across various sample types
Three different lots of the Sequential Protein/DNA/RNA Extraction Kit were evaluated across multiple biologically relevant sample types, including HeLa cells, PBMCs, and mouse liver tissue, using inputs of 100,000 HeLa cells, 500,000 PBMCs, and 2 mg of tissue per isolated sample. Each sample type was tested in triplicate for each reagent lot. As shown in Figure 11, the three independent kit lots generated highly consistent protein, DNA, and RNA yields across all sample types, demonstrating strong lot-to-lot reproducibility and reliable performance across diverse materials.
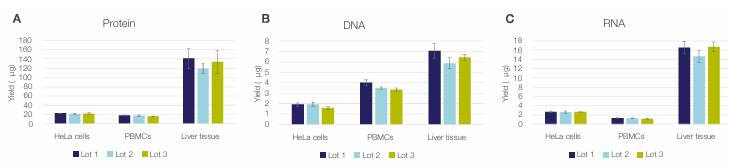
Figure 11. Reproducibility of the Sequential Protein/DNA/RNA Extraction Kit workflow. Yields are of (A) protein, (B) DNA, and (C) RNA extracted
from 100,000 HeLa cells, 500,000 PBMCs, and 2 mg mouse liver tissue using three different reagent lots are shown. Error bars represent the standard
deviation of three technical replicates (n = 3).
Conclusion
The Sequential Protein/DNA/RNA Extraction Kit helps deliver efficient and versatile multiomics sample preparation by enabling sequential isolation of protein, DNA, and RNA from a single sample. When paired with the KingFisher Apex, Flex, or Duo Prime sample purification systems, it offers a robust, automation-ready workflow that reduces hands-on time, increases throughput, and conserves precious or limited specimens. Sequential extraction of proteins, DNA, and RNA from the same sample does not reduce yield or integrity for any analyte compared with splitting the sample and using separate single-analyte kits (data not shown).
By replacing traditional manual, column-based methods with magnetic bead–based technology, the kit streamlines the workflow and maintains analyte consistency, supporting downstream mass spectrometry for proteins and next-generation sequencing, qPCR, and RT-qPCR for nucleic acids. Its flexibility across a wide input range (1,000–1,000,000 cells or 2.5 µg–10 mg of tissue) and compatibility with diverse sample types make it a powerful tool for multiomics research.
This multiomics workflow enables sequential extraction of protein, DNA, and RNA from limited or precious samples, facilitating integrated proteomic, genomic, and transcriptomic analyses. The automatable protocol reduces hands-on time and scales to high throughput, thereby helping to support biomarker discovery, advance precision medicine, and accelerate development of liquid biopsy assays.
In summary, the combination of sequential multi-analyte recovery and scalable automation makes the Sequential Protein/DNA/RNA Extraction Kit an excellent solution for enhancing productivity, minimizing variability, and generating high-quality protein, DNA, and RNA for multiomics research.
References
1. Woodland B et al. (2025) Sample preparation for multi-omics analysis: considerations and guidance for identifying the ideal workflow. Proteomics 25:76–101.
2. Gao F et al. (2024) Comparative analysis of high-throughput RNA extraction kits in naïve non-human primate tissues. Anal Biochem 684:115293.
3. Distler U et al. (2022) Protein extraction and sample preparation strategies for shotgun proteomics of CNS cells and brain tissue. Methods Mol Biol 2361:1–18.
4. Frangogiannis NG (2019) The extracellular matrix in ischemic and non-ischemic heart failure. Circ Res 125:117–146.
5. Molden RC et al. (2011) Challenges in liver tissue nucleic acid isolation. BioTechniques 51:279–286.
6. Ausubel FM et al. (eds.) Current Protocols in Molecular Biology. Wiley (updated continuously).
7. Tabb DL et al. (2010) Repeatability and reproducibility in proteomic identifications by liquid chromatography–tandem mass spectrometry. J Proteome Res 9:761–776.
8. Shang R et al. (2023) microRNAs in action: biogenesis, function and regulation. Nat Rev Genet 24:816–833.
9. Sato F et al. (2011) MicroRNAs and epigenetics. FEBS J 278:1598–1609.
10. Croce CM (2009) Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10:704–714.
11. Mitchell PS et al. (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 105:10513–10518.
12. Park JE et al. (2021) The roles of the let-7 family of microRNAs in regulation of cell proliferation, differentiation and cancer. Cells 10:2415.
13. Cimmino A et al. (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 102:13944–13949.
 Kit format: The Sequential Protein/DNA/RNA Extraction
Kit format: The Sequential Protein/DNA/RNA Extraction
Kit comes with 12 individual components in a space-saving
design for convenient storage suitable for 24 or 100 reactions.
The 100-reaction vials can also be purchased as separate
components. All components of the kit are clearly labeled, and
color-coded caps (orange, green, blue, white) indicate protein,
DNA, RNA, or all isolation.
Storage conditions: All reagents are stored at room temperature
(15–25°C), except the Invitrogen™ Dynabeads™ Sequential
Protein Binding Beads and the Sequential Protein Elution Buffer,
which need to be stored at 2–8°C.